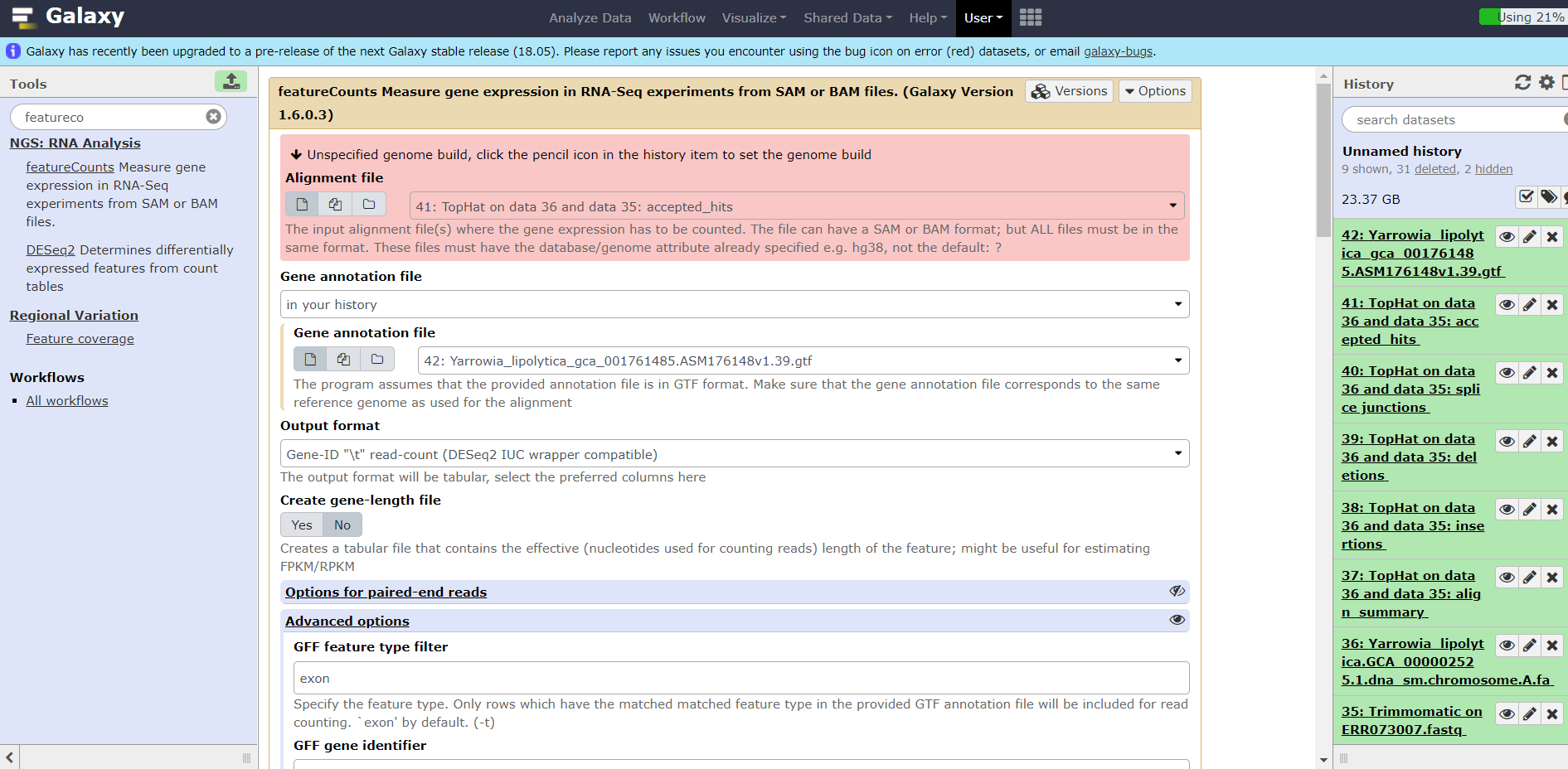
I have a trouble with using a data of an experiment about yeast Yarrowia lipolytica. Firstly ı could't get the file from the site www.ebi.ac.uk, so ı have downloaded the file to my pc and the uploaded the file to galaxy. Then I have used succesfully top hat and trimmomatic tools and succesfully loaded fasta and gtf formatted files about Yarrowia lipolytica. But at the end when ı wanted to use the tool "Featurecounts", a warning appears and says "can not use unspecified species, please click the pencil icon and change the species." The obtained file format was bam, but name of species was undefined. I did click this icon but ı couldn't select the species name because there was no loaded data about Yarrowia lipolytica, and also there was no option to load additional files of Yarrowia. So ı could't change or load any information about Yarrowia Could you help me please. Sincerely yours,
Mehmet SOLAK PhD student Yeast Genetics Laboratory/Department of Biology University of 18 MART / CANAKKALE / TURKEY mserafeddinsolak@gmail.com